This website is intended for healthcare professionals only.
Take a look at a selection of our recent media coverage:
8th November 2024
Moving medical supplies via drone was once a farfetched idea, but there are now multiple projects working to make it a reality. Kathy Oxtoby considers five case studies from the UK and the Netherlands to determine the benefits for clinicians, patients and healthcare systems, as well as the remaining challenges and future potential of drone transportation within the healthcare landscape.
With rising waiting lists, ongoing staff shortages and mounting pressure on hospital teams, it is increasingly vital that care is delivered faster and more efficiently to patients. Greener, more sustainable care is also a priority, with the drive to achieve a net zero NHS by 2040.
One approach that has the benefits of both faster delivery and sustainability is the use of drones – also known as unmanned arial vehicles or UAVS. Drones are already transporting medicines to remote areas in such countries as Rwanda, the United States, Australia and India.
In recent years, the NHS has been trialling the use of drones for a variety of purposes, including delivering medical supplies of blood packs and chemotherapy, transporting lab specimens, and more.
For Professor Claire Anderson, Royal Pharmaceutical Society president, the Covid-19 pandemic and recent advancements in technology have made the drone transportation of medical supplies eminently possible, and the benefits are clear.
‘Drones offer timely access to medicines, especially in remote areas, and the pandemic highlighted their potential for safe, contactless delivery of essential supplies,’ she says.
‘It can reduce costs and travel time, improve access to healthcare for patients in rural or hard to reach areas, and free up staff time for direct patient care. Drones are also more environmentally sustainable, emitting less carbon dioxide than cars or trucks.’
For patients with chronic conditions, using drones to deliver critical medicines can ‘help reduce waiting times and ensure more consistent access to healthcare’ and ‘alleviate some of the pressures caused by long waiting lists’, she adds.
However, in their current form, there are some inevitable drawbacks. Drones can only carry light items – typically around two to four kilograms – which limits their use for transporting a wide range of items. Additionally, they have limited range and battery life, which affects the numbers of deliveries that can be made on one flight.
In addition, some medicines need to be stored in specific conditions, such as controlled temperatures and multilayer packaging, which must also be taken into consideration. ‘Regulations require these conditions to be met throughout transport to ensure the product is safe to use,’ says Professor Anderson.
As the need and momentum for the use of drones in healthcare builds, the list of projects assessing the benefits, challenges and future potential of drone transportation is growing.
So what projects are currently underway, and what insights are they giving into this innovative movement of medical supplies?
The weather and geography of Cornwall, southwest England, present unique challenges when it comes to the collection and delivery of pathology samples and time-critical medicines, particularly on the Isles of Scilly – 28 miles off the coast.
Poor weather conditions mean flights to and from the islands can be grounded for two to three days, delaying transit of crucial items.
The Open Skies Cornwall project is a consortium of technology providers and end users. It includes exploring conceptual use cases involving the transport of pathology samples and blood products, point of care equipment and consumables, and Royal Cornwall Hospital Pharmacy service provision via drones.
‘We wanted to level up the provision of care and build a reliable and robust service for the island community,’ says Jo Walsh, pathology optimisation project lead at Royal Cornwall Hospitals NHS Trust.
Samples can be adversely affected by the time it takes to transport them, and often repeat testing is required as a result. ‘We’re looking to prevent any repeated testing and provide timely and accurate results for clinicians, that are not adversely affected by transport delays,’ says Ms Walsh.
‘We also want to enable patients to receive treatment at home, rather than having to travel to the main hospital – a journey that is especially problematic for those living off the mainland, as they have to travel by plane.’
As well as focusing on island healthcare connectivity, the Open Skies Cornwall project also involves working with Falmouth Harbour to integrate autonomous drone solutions and enable infrastructure for ship-to-shore delivery, remote healthcare, telemedicine and flying defibrillator applications to support residents and maritime visitors.
Lisa Vipond, pathology services manager at the Trust says that the team sees ‘this project as a complimentary element to our courier system, assisting their challenges.’
A key part of the project is looking at maintaining the validity of samples, and the impact of environmental factors, such as heat, cold, pressure and vibrations on samples transported by drone.
There are plans to do testing flights at the end of this year, but ‘we need to ensure the regulatory and legislative requirements are all in place ahead of this’, says Ms Vipond.
Ms Walsh believes many other areas of the NHS could benefit from drones, including emergency care.
‘Working in the NHS, patient care is at the centre of what you do,’ says Ms Walsh. ‘When you see a solution to gaps in service provision due to elements beyond your control – such as geographical and weather limitations – you want to push that solution forward.
‘We can’t just use this project as a “proof of concept”. We need to embed drone transport within our infrastructure long-term.’
Project CAELUS (Care & Equity – Healthcare Logistics UAS Scotland), aims to develop and trial the UK’s first national distribution network using drones to transport essential medicines, blood, organs and other medical supplies throughout Scotland to eliminate land transport issues.
Led by AGS Airports, it brings together 16 partners, including NHS Scotland and is funded by the UK Research and Innovation Future Flight Challenge and other partners. The consortium has developed a virtual model, or digital twin, of the proposed delivery network, which connects hospitals, pathology laboratories, distribution centres and GP surgeries across the country.
A number of live flight trials are taking place across the country as part of the project. For example, this August, laboratory specimens were flown between NHS Lothian and NHS Borders by drone.
In October, drone technology was used to connect the island community of Arran with the mainland. Further trials are planned in the NHS Highland and NHS Grampian areas of Scotland later in the year.
And the Scottish Ambulance Service has also researched whether a drone could transport defibrillators to the location of a cardiac arrest faster than an ambulance.
Dr Jamie Hogg, clinical lead for Project CALEUS for the north of Scotland and a retired GP, says the team hopes that the use of drones to transport medicines, blood samples and equipment will enable patients living in more rural areas to be ‘treated closer to home and more quickly’.
Requesting quick deliveries of medicines for patients via a drone network would have significant benefit, he says, however, a change in regulations to allow the move from the currently segregated to integrated airspace will be key.
The project ends in December, and then there will be ‘a period of reflection to take in everything that’s been done and decide on next steps’, Dr Hogg explains.
Blood packs have been successfully flown by drone in a series of ‘beyond visual line of sight’ flights, for the first time in the UK.
In a research study to check the viability of flying blood via drone, run jointly by NHS Blood and Transplant (NHSBT) and the medical logistics company Apian, 10 units of packed blood cells were transported on a 68km journey across Northumbria’s skies, while an identical 10 packs were transported concurrently by road.
After assessment, results showed both sets remained viable, with no significant difference in the biochemical or haematological profiles of the blood, which determine if it has maintained quality and can be used for clinical purposes.
‘We’re proud to drive innovation that could improve patient outcomes, and this trial could do exactly that,’ says Mike Wiltshire, component development laboratory manager at NHS Blood and Transplant.
‘Drone travel would be especially useful in transporting items – whether blood packs, blood samples or other – to more remote locations, or via routes that normally suffer from traffic congestion, meaning the products are available for patients faster than they would be by road and ensuring patients are treated as quickly as possible.’
If drones are able to deliver blood products faster, then ‘more patients will be able to be treated or receive results the same day’ than at present, which ‘may reduce patients having to return to the hospital at a different time, should the medicine or test results not be available same day’, Mr Wiltshire adds.
The UK has clear guidelines on the transport of blood components and maintenance of product temperature. ‘We needed to source a suitably sized and specified transport container, along with cool packs, to ensure the temperature of the blood was maintained as required,’ explains Mr Wiltshire.
The number of items and weight that can be transported at any one time is limited by the drone load capacity. Drone operators are therefore exploring different types of drone to determine the best one for the transportation of blood, which may in turn be dependent on the specific requirements of the transport route.
The flying of drones like those used in this study is currently ‘very tightly regulated’ meaning that drones cannot simply fly directly between any given two points – permission must be granted, which may not be guaranteed, depending on the locations in question. ‘Drone operators are looking to overcome this significant challenge for the use of drones for this and many other uses,’ he says.
NHSBT is currently in discussions around a similar trial for platelets, to understand how platelets for transfusion will react to drone transportation and whether their use will be viable in the NHS for this purpose.
The Welsh Blood Service (WBS) is interested in exploring what role drones might play in enabling efficient, sustainable transport of blood products between north and south Wales as well as faster, on-demand delivery of blood products and other medical supplies in rural Wales.
The organisations involved in the Welsh Health Drone Innovation Partnership are the WBS – part of Velindre University NHS Trust –the Welsh Ambulance Service University NHS Trust, Snowdonia Aerospace Centre and the technology company Slink-Tech.
Currently, the partnership is undertaking a foundation study for drone-based blood delivery service between WBS stock holding units at Talbot Green in the south and Wrexham in the north to establish its potential for supporting the Welsh NHS, including specific use cases for the WBS, and to test the basic premise with the Civil Aviation Authority.
‘Drone-based infrastructure has the benefit of not being tied to pre-existing infrastructure on land, which due to geographical constraints has often unintentionally left rural communities underserved,’ says a spokesperson for Velindre University NHS Trust.
‘Drone technology provides the opportunity to tackle inequalities by improving accessibility to communities and regions which may be left behind by traditional logistic infrastructure.’
The primary challenge is to establish ‘a robust business case for early deployment of drone technology to improve the quality and resilience of health and care services in Wales’, the spokesperson adds.
Alan Prosser, the director of the WBS, says: ‘Technology is advancing at pace in this area, and we acknowledge that drone capability still needs to mature in terms of carrying capacity and battery payload before this becomes a viable option for our service.’
The UK isn’t the only country trialling the use of drones to transport medical supplies. In the Netherlands, researchers have investigated the impact of medical drone transport on the stability of monoclonal antibodies (mAbs).
The study findings revealed ‘no significant differences between car and drone transport’, indicating that the stability of mAbs in both vials and infusion bags was adequately maintained during transportation regardless of the mode.
As such, medical drones are ‘a viable and reliable means for the inter-hospital transport of mAbs, paving the way for more efficient and predictable logistics in healthcare delivery’, the authors say.
In fact, the researchers concluded that the integration of drone technology into healthcare logistics ‘has the potential to significantly enhance’ the crucial transport of this treatment type.
With so many ongoing trials and success stories demonstrating the benefits of drone technology in healthcare, the future looks bright, and Professor Anderson says it really does have ‘the potential to ‘revolutionise the way we deliver medical supplies, especially in remote or hard-to-reach areas’.
She is keen to point out, however, that ‘as with any transport around medicines, safety and security must remain a priority’.
The use of drones will ‘undoubtedly increase over the next five to 10 years, for a variety of applications,’ according to Mr Wiltshire. ‘However, there are challenges to overcome – such as restrictions on airspace – before this use is widespread’.
In the meantime, Dr Hogg is encouraging healthcare professionals to ‘think about what they could do if they had drones available to them’ to support patients in accessing vital medical supplies.
‘We are getting to the point where drone transport for medical products could become a reality,’ he says. ‘In three- or four-years’ time, we could be saying to a [resident] doctor: “Can you “drone” this down to Aberdeen?”, and the answer will be: “Yes, sure.”’
15th August 2024
A research team from the Netherlands has found that the stability of monoclonal antibodies (mAbs) is unaffected by the use of drone transportation when compared with traditional car transport for inter-hospital transportation.
They found the stability of mAbs in both vials and infusion bags was adequately maintained during drone transport, indicating that medical drones are a viable and reliable transportation method.
The current reliance on cars to deliver mAbs is compromised by the unpredictable nature of traffic and infrastructure, which leads to unreliable transport times. Drone transport could therefore offer health systems a more efficient and predictable transportation system for mAbs, the authors said.
The researchers assessed the efficacy and safety of medical drone transportation on the stability of mAbs transported in vials and ready-to-administer infusion bags with blinatumomab, tocilizumab and daratumumab. These were Blincyto 38.5 μg concentrate after reconstitution, RoActemra 20 mg/mL concentrate for infusion and Darzalex 20 mg/mL concentrate for infusion, respectively.
These mAbs were chosen as blinatumomab represents a low-concentrate protein drug, tocilizumab represents a drug that may be necessary in emergency settings, and daratumumab is a viable option for home treatment of patients.
Using a VTOL fixed-wing drone, temperature recorders and impact indicators estimated mechanical stress during the flight. Four flights carrying vials and five flights carrying IV bags were performed at a field lab in the Netherlands with an average flight length of eight kilometres and eight minutes of flight time.
Control high-performance size-exclusion chromatography (HP-SEC), dynamic light scattering (DLS), light obscuration (LO), micro-flow imaging (MFI) and nanoparticle tracking analysis (NTA) and absence of visible particles (VI) were employed to assess the presence of aggregates and particle formation in car, drone and control conditions.
Where there was enough quality data, statistical analysis of the vials revealed no significant differences between the control group and the drone-exposed mAbs. Similarly, there were no statistically significant differences between the control, the car and the drone groups for all the infusion bags. By way of example, the data for blinatumomab has been summarised in Table 1 below.
Table 1: Summary of the main results of infusion bag analysis
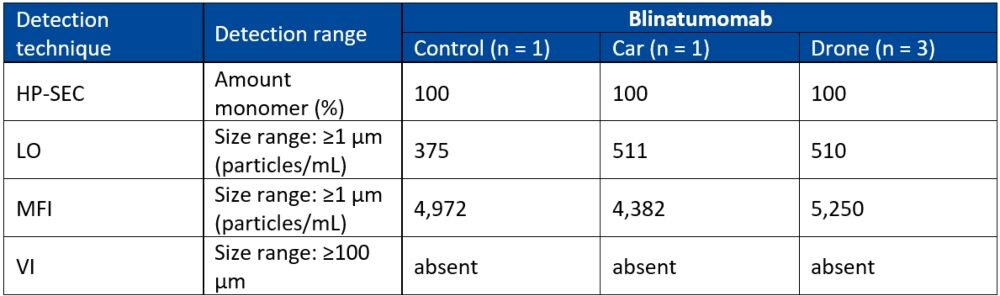
HP-SEC: mean percentage, DLS: mean particle size, LO and MFI: mean total particle concentration, VI: absence of visible particles.
The researchers concluded that integrating drone technology into healthcare logistics has the potential to significantly enhance the efficiency of inter-hospital transportation. Both commercial vials and ready-to-administer infusion bags of mAbs could be transported by drone without resulting in aggregate formation.
Güngören M et al. Investigating the Impact of Drone Transport on the Stability of Monoclonal Antibodies for Inter-Hospital Transportation. Journal of Pharmaceutical Sciences 2024; Apr 03: doi.org/10.1016/j.xphs.2024.04.002.
13th April 2023
Dupilumab treatment for 12 months leads to the lowest number of exacerbations and better improvements in FEV1 compared to two commonly used monoclonal antibodies (mAbs), omalizumab or mepolizumab, in adults with moderate to severe asthma, according to comparative analysis by US researchers.
The prevalence of severe asthma varies between 3.6 and 6.1% of all patients with the disease. Individuals with moderate to severe asthma have the opportunity of being treated with several monoclonal antibodies, which target key inflammatory cytokines involved in disease pathogenesis. All of these currently approved biologic therapies have been shown to improve asthma-related outcomes in individuals with asthma uncontrolled with conventional therapy. However, for patients with more than one phenotype such as allergic and eosinophilic asthma, trying to identify the most suitable agent is less clear. In the absence of direct head-to-head comparative trials, researchers can often utilise observational data to emulate a hypothetical pragmatic randomised trial, which is referred to as the target trial.
In the current analysis, the research team emulated a hypothetical randomised trial, making use electronic health records from a large US-based academic health care system. They included adult participants with baseline IgE levels between 30 and 700 IU/mL and peripheral eosinophil counts of at least 150 cells/μL with the objective of comparing the effectiveness of dupilumab treatment with omalizumab or mepolizumab in patients with moderate to severe asthma. The main outcomes of interest were the incidence of asthma-related exacerbations and the change in baseline FEV1 value over 12 months of follow-up.
Dupilumab treatment and asthma outcomes over 12 months
In all, 68 individuals received dupilumab treatment 68 received omalizumab and 65 received mepolizumab.
Over the 12 month period of follow-up, asthma-related exacerbations occurred in 25.0% of those receiving dupilumab treatment compared to 43.1% for mepolizumab (adjusted hazard ratio, aHR = 0.35, 95% CI 0.18 – 0.71). Compared to omalizumab group, asthma exacerbations occurred in 39.7% of patients, giving a corresponding adjusted hazard ratio of 0.42 (95% CI 0.20 – 0.87).
The change in FEV1 for the different agents were 0.11 L (95% CI -0.003 to 0.222 L) for dupilumab versus mepolizumab and 0.082 L (95% CI -0.040 to 0.204 L) for dupilumab versus omalizumab.
In patients with eosinophil counts of at least 300 cells/μL, the HR for dupilumab compared to mepolizumab was 0.26 (95% CI 0.10 – 0.67) and 0.24 (95% CI 0.09 – 0.63) for dupilumab vs omalizumab.
Based on these findings, the authors concluded that among patients with asthma and eosinophil counts of at least 150 cells/μL and IgE levels of 30 to 700 kU/L, dupilumab was associated with greater improvements in exacerbation and FEV1 value than either omalizumab or mepolizumab.
Citation
Akenroye AT et al. Comparative effectiveness of omalizumab, mepolizumab, and dupilumab in asthma: A target trial emulation. J Allergy Clin Immunol 2023
25th March 2022
Tixagevimab/cilgavimab (Evusheld) is a combination of two monoclonal antibodies that has been approved by the UK’s MHRA for use before exposure to COVID-19 in order to prevent the disease.
The drug would therefore be most suited to adult patients who are unable to mount a sufficient immune response after receiving a COVID-19 vaccination or alternatively, patients for whom vaccination is not recommended.
However, the combination would not be suitable for those currently infected with COVID-19 or who have had a recent and known exposure to someone infected with the virus. The two components of Evusheld are available as separate intramuscular injections and research has shown that these recognise non-overlapping sites and are simultaneously bound to the S protein and neutralise the wild-type COVID-19 virus in a synergistic manner.
As a result, the manufacturer, AstraZeneca, has examined the value of Evusheld in three separate clinical studies.
Evusheld clinical efficacy
To date, none of the three major clinical studies have been fully published and the efficacy data has been made available in press releases from the manufacturer. Evusheld was examined in the PROVENT trial which was designed to assess the safety and efficacy of a single dose compared to placebo for the prevention of COVID-19.
The trial included 5,197 participants and who were randomised 2:1 to a single 300 mg dose of Evusheld (AZD7442 in all press releases) or placebo and which was administered in two separate, sequential IM injections. The trial recruited individuals 18 years of age and over (including 43% who were older than 60 years of age) who would benefit from prevention and were defined as having an increased risk for an inadequate response to active immunisation or having an increased risk for COVID-19 infection.
Participants at the time of screening were unvaccinated and had a negative COVID-19 test. The primary efficacy endpoint of the trial was the first case of any COVID-19 PCR confirmed, symptomatic illness occurring after the dose before day 183.
According to a press release from the manufacturer, Evusheld reduced the risk of developing symptomatic COVID-19 by 77% (95% CI 46 – 90%) in comparison to those given a placebo.
A second trial, STORM CHASER, was designed to explore post-exposure prophylaxis of COVID-19 in Adult patients. The trial included 1,121 participants, randomised 2:1 as before to either Evusheld or placebo, all of whom tested negative for COVID-19 prior to receiving treatment.
Again, in a press release from the manufacturer, Evusheld reduced the risk of developing symptomatic COVID-19 by 73% (95% CI 27 – 90%) compared with placebo among those who were PCR negative at the time of dosing.
The third trial, TACKLE, explored the value of Evusheld given to adults who were non-hospitalised with mild-to-moderate COVID-19 and symptomatic for seven days or less, but this time, given a 600 mg dose of the drug.
The primary efficacy endpoint of the trial was the composite of either severe COVID-19 or death from any cause through day 29.
According to the manufacturer press release on TACKLE, Evusheld given to participants within five days of symptom onset, saw a 67% reduced risk of developing severe COVID-19 or death compared to placebo.
On the basis of these findings, the MHRA has approved the drug and in Europe, the EMA is currently evaluating the combination.
19th January 2022
Casirivimab and imdevimab used together lead to a significant reduction in the proportion of patients who develop symptomatic COVID-19 compared to those given placebo. This was the conclusion of a randomised trial by researchers from the manufacturer of the combination, Regeneron Pharmaceuticals, New York, US.
Although the arrival of effective COVID-19 vaccines have been shown to boost individuals’ immunity against the virus, therapy with monoclonal antibodies remains an alternative for those who develop an inadequate response to vaccination.
However, a recent Cochrane systematic review of monoclonal antibody therapy in COVID-19, concluded that ‘our certainty in the evidence for all non‐hospitalised individuals is low, and for hospitalised individuals is very low to moderate. We consider the current evidence insufficient to draw meaningful conclusions regarding treatment with SARS‐CoV‐2‐neutralising mAbs‘ (monoclonal antibodies).
Casirivimab and imdevimab are two mAbs that bind to different parts of the spike protein present on the surface of COVID-19. Furthermore, trial data has revealed how this combination reduced the risk of COVID-19–related hospitalisation and all-cause mortality, as well as resolving symptoms and decreasing viral load, more quickly than placebo.
The Regeneron Pharmaceuticals team undertook a two phase study of their combination. In part A, casirivimab and Imdevimab were found to prevent both symptomatic and asymptomatic COVID-19 infection among previously uninfected household contacts of infected individuals.
The present study relates to part B of their trial, in which asymptomatic, infected close contacts were treated with subcutaneous casirivimab and imdevimab.
Part B was a randomised, double-blind, Phase III trial, designed to determine whether subcutaneous casirivimab and imdevimab could prevent progression from asymptomatic to symptomatic infection.
Enrolled participants were adults with a PCR confirmed COVID-19 infection, identified within 96 hours of another household contact testing positive. Included participants were then randomised 1:1 to casirivimab and imdevimab or placebo and the primary endpoint of the trial was the proportion of individuals who developed signs and symptoms of COVID-19 within 14 days of their positive PCR result and this was reviewed over a 28 day period following randomisation.
Findings
A total of 314 individuals with a mean age of 41 years (51.6% female) were included of whom, 204 (66%) were asymptomatic and randomised to either casirivimab and Imdevimab (100) or placebo.
Among asymptomatic individuals assigned to treatment, 29% became symptomatic compared to 42.3% of those given placebo (odds ratio, OR = 0.54, 95% CI 0.30 – 0.97, p = 0.04).
Participants on treatment also experienced a 5.6 day reduction in the mean duration of symptoms compared to placebo and the total number of weeks with a high viral load was significantly reduced (489.9 weeks vs 811.9 weeks per 1000 participants, treatment vs placebo, p = 0.01).
The authors concluded that treatment with casirivimab and imdevimab for asymptomatic COVID-19 positive individuals living with an infected household contact, significantly reduced the incidence of symptomatic infection over a period of 28 days.
Citation
O’Brien MP et al. Effect of Subcutaneous Casirivimab and Imdevimab Antibody Combination vs Placebo on Development of Symptomatic COVID-19 in Early Asymptomatic SARS-CoV-2 Infection. A Randomized Clinical Trial JAMA 2022.
31st August 2021
Urticaria is characterised by the presence of pruritic wheals (i.e., hives), angioedema and in some cases both. In the majority of cases, urticaria resolves within two days though when it persists for longer than six weeks, the condition is referred to as chronic spontaneous urticaria (CSU). The prevalence of CSU varies across the world and one recent systematic review found a prevalence of 1.4% among Asians but only 0.1% in people from North America. All forms of urticaria are mast cell-related diseases and the symptoms due to the release of histamine and other cytokines. The condition can be manged through the use of H1 anti-histamines but evidence shows that in CSU, antihistamines at licensed doses, are effective in less than half of all patients. Other therapeutic options under investigation include monoclonal antibodies such as omalizumab which has been approved for the management of CSU although the relative effectiveness of monoclonal antibodies in antihistamine-resistant CSU is uncertain.
This led a team from the Department of Pharmaceutical care, Chiang Mai University, Thailand, to undertake a systematic review and network meta-analysis, of pharmacological therapies in antihistamine-refractory CSU. The team looked for randomised trials in both adolescents and adults with diagnosed antihistamine-refractory CSU and which had used a validated measurement tool for treatment assessment. The researchers set the primary outcome as a change in urticaria symptoms, both hives and pruritus from baseline but also considered the unacceptability of treatment in terms of the number of patients dropping out. The outcomes were reported as standardised mean differences (SMD).
Findings
There were 23 randomised trials of SCU with 2480 patients and a mean age ranging from 32.2 to 43.8 years. The trials compared a total of 28 different interventions and at licensed antihistamine doses and up-dosed (i.e., where the antihistamine dosage was increased by two to four times the licensed dose). The most effective treatment, with a large effect size, was the monoclonal antibody ligelizumab at a dose of 72 mg (SMD -1.05, 95% CI -1.37 to – 0.73) and at the higher dose of 240 mg (SMD – 1.07, 95% CI -1.39 to – 0.75). Omalizumab produced a moderate effect size at a dose of 300 mg (SMD – 0.77, 95% CI -0.91 to -0.63). There were no significant differences in treatment unacceptability.
In their discussion, the authors suggested that either ligelizumab at a dose of 72 or 240 mg and omalizumab 300mg were the most effective treatments for refractory chronic spontaneous urticaria. In addition, they called for head-to-head trials to provide improved estimates for the effectiveness of these treatments.
Citation
Nochaiwong S et al. Evaluation of Pharmacologic Treatments for H1 Antihistamine–Refractory Chronic Spontaneous Urticaria. A Systematic Review and Network Meta-analysis. JAMA Dermatol 2021
24th August 2021
Psoriasis is best described as a complex, chronic, multifactorial and inflammatory disease characterised by increased proliferation of keratinocyte cells in the skin giving rise to silvery/white plaques. The disease typically presents on extensor surfaces such as the elbows, knees and lower back and there if often involvement of the scalp. The incidence of psoriasis appears to vary across the world, with a recent study noting a wide variation. For instance, the incidence was 30.3 per 100,000 person years in Taiwan but 321 per 100,000 person years in Italy.
Among those with psoriasis, one study of over 9,000 patients, found that just over half (51.8%) had mild disease, with 35.8% and 12.4% having moderate and severe disease respectively. Patients with mild to moderate disease are normally managed with topical therapies, whereas those with moderate to severe disease are increasing treated with biological agents, in particular, monoclonal antibodies. Although the precise cause of psoriasis remains to be determined, several interleukins appear to be important disease drivers, especially the interleukin-17 (IL-17) pathway which includes six similar agents, IL-17A – IL-17F.
Research suggests that two members of the IL-17 family, IL-17A and IL-17F are implicated in autoimmunity and IL17F appears to regulate pro-inflammatory gene expression and which requires the IL-17A receptor, suggesting that both are involved in the pathology of psoriasis. Bimekizumab is the first monoclonal antibody which selectively inhibits both IL-17A and IL-17F and is therefore a potentially important advancement in the management of patients with moderate to severe psoriasis.
Clinical efficacy
The EU approval of bimekizumab was supported by the results of three phase 3 clinical trials, all undertaken in patients with moderate to severe psoriasis. The first, BE READY, randomised 435 patients (4:1) to bimekizumab 320 mg every 4 weeks or placebo. The co-primary endpoint was a PASI90 (i.e., 90% improvement in disease severity compared to baseline) and the proportion of patients achieving a score of 0 (i.e., clear skin) or 1 (almost clear), based on a 5-point investigator global assessment (IGA) score after 16 weeks of treatment.
The results showed that a staggering 91% of those assigned to bimekizumab achieved a PASI90 compared to only 1% in the placebo group. In the BE VIVID trial, 567 patients were randomised to bimekizumab (at the same dosage as the BE READY trial) or this time, ustekinumab 45 or 90 mg every 12 weeks and which is an active comparator or placebo. Once again at week 16 there was a high response in the bimekizumab group with 85% achieving a PASI90 compared to 50% in the ustekinumab. The third trial, BE SURE, enrolled 478 patients who were randomised to either bimekizumab (same dosage as in the other trials) or adalimumab (another active comparator) at a dose of 40 mg every 2 weeks. At week 16, a PASI90 was achieved 85.3% of those using bimekizumab and 57.2% of those given adalimumab.
The most common adverse effects from bimekizumab are upper respiratory tract infections (14.5%) and patients are advised to seek medical advice if they display symptoms suggestive of an infection.
The EU approval applies to all 27 member states as well as Iceland, Liechtenstein and Norway at a dose of 320 mg administered by subcutaneous injections every 4 weeks for week 16 and every 8 weeks thereafter. The drug is currently under review by the US Food and Drug administration.
6th August 2021
Systemic lupus erythematosus (SLE) is a rare, chronic autoimmune disease which affects approximately six-times more women than men. It is the most common form of lupus, accounting for approximately 70% of all cases. There is a wide geographical variation in the incidence of SLE and has been found to vary between 23.2 cases per 100,000 in North America to 0.3 cases per 100,000 in Africa and the Ukraine. Other estimates suggest a general prevalence of approximately 1 to 10 per 100,000 person-years.
SLE is a systemic illness affecting many different organ systems including the skin, with a typical butterfly rash across the cheeks, the musculoskeletal system, producing arthritis and myositis, and constitutional symptoms such as fatigue, fever and weight loss. The cause of SLE remains unclear but the condition is characterised by the presence of autoantibodies and guidance from EULAR in 2019 has suggested treatment with hydroxychloroquine, glucocorticoids, immunosuppressants and biologics, in particular, belimumab. However, it has been shown that in patients with SLE, there is excessive production of type 1 interferon (IFN) and in particular, INF-alpha. Blockage of type 1 IFN could therefore represent a potential therapeutic modality for SLE.
Anifrolumab
Cell signalling in SLE occurs via activation of the IFN pathway mediated via the type 1 IFN receptor. Moreover, blockage of this IFN pathway may reverse immune dysregulation and the tissue damage seen in SLE. Anifrolumab binds to the IFN receptor and therefore potentially reduces some of the immune dysregulation seen in SLE.
The FDA based its approval of anifrolumab on the results of three separate trials. The first in 2016, undertaken in 305 patients, showed that treatment with intravenous anifrolumab (300mg or 1000mg) every 4 weeks for 48 weeks led to a substantial reduction in disease activity in patients with moderate-to-severe SLE. Further studies included TULIP-1 and TULIP-2, both of which were Phase III trials. The primary endpoint was the rate of British Isles Lupus Assessment Group-based Composite Lupus Assessment (BICLA), which is a validated global measure of treatment response among those with SLE. Interestingly, anifrolumab failed to reach the BICLA endpoint in TULIP-1 (37% vs 27%, anifrolumab vs placebo) when added to standard therapy. In contrast, significantly more patients assigned to anifrolumab than placebo, achieved the primary endpoint in TULIP-2, (47.8% vs 31.5%, anifrolumab vs placebo, p = 0.001).
An ongoing clinical trial in approximately 360 participants with moderate-to-severe SLE, is currently assessing the efficacy and safety of a subcutaneous form of anifrolumab in adults receiving standard therapy, over a 52-week period.
Anifrolumab is under regulatory review in both the EU and Japan.
Source. AstraZeneca August 2021
14th July 2021
Retrospective real-world data confirms the effectiveness of the monoclonal antibodies mepolizumab and benralizumab in eosinophilic asthma.
Eosinophilic asthma is characterised by increased eosinophilic inflammation, recurrent exacerbations and poor disease control. The proportion of asthmatics with the eosinophilic phenotype varies with one study suggesting a prevalence of between 21% and 69% in adults.
However, other work has suggested that while the prevalence is potentially less than 1%, asthma-related healthcare resource use among those with eosinophilic asthma was four times greater than for other phenotypes.
A further complication from eosinophilic asthma is that lung remodelling and airway hyperinflation leads to a persistent decrease in pulmonary function. Eosinophilic asthma is also associated with over-expression of Th2 inflammatory cytokines such as interleukin-5, which is known to play an important role in the recruitment and survival of eosinophils in peripheral tissues.
Two monoclonal antibodies approved for the treatment of severe eosinophilic asthma are mepolizumab and benralizumab. Mepolizumab binds to and inactivates interleukin-5 and in patients with severe asthma, requiring daily oral glucocorticoid therapy, mepolizumab has been shown to reduce disease exacerbations, improve symptom control and produce a steroid-sparing effect.
Similarly, benralizumab, which targets the interleukin-5 receptor alpha subunit, depleting eosinophils, has also been found to improve symptoms of severe asthma which remains uncontrolled by high-dosage inhaled corticosteroids and long-acting beta-agonists. While both monoclonal antibodies are effective, what remains unclear is whether there are any important clinical differences between the two agents.
This prompted a team from the Department of Respiratory Medicine, Hannover Medical School, Munich, Germany, to undertake a retrospective analysis of the comparative efficacy of the two monoclonal antibodies in severe asthma.
Included patients were those with a physician diagnosed severe eosinophilic asthma, which despite the use of high-dose inhaled glucocorticoids and long-acting beta-agonists, plus optional second or third-line control, and/or oral glucocorticoids, did not provide sufficient control.
For such patients, the treating physician, decided to use either mepolizumab 100mg or benralizumab 30mg every four weeks for the first three doses and then once every eight weeks. The researchers focused on clinical factors such as asthma control, pulmonary function, exacerbation rates, oral glucocorticoid use over a 12-month period. Symptom data were collected at baseline and after six and 12 months.
A total of 187 patients, 123 given mepolizumab, were included in the analysis. The median age was 58 years (42% female) and 54% were described as non-smokers. Use of both monoclonal antibodies led to a significant improvement in pulmonary function as defined by changes in the forced expiratory volume in one second (FEV1). This increased from 59% to 74% for mepolizumab and from 63% to 72% for benralizumab.
The asthma control scores also significantly improved for both drugs: 13 to 19 (p < 0.001) for mepolizumab and 12 to 21 (p < 0.001) for benralizumab when assessed after six months. The number of exacerbations also reduced with 68% of patients in either group reporting no exacerbations at all during the 12-month follow-up period and there was a significant reduction in the dosage of oral glucocorticoids used.
The authors concluded that both monoclonal antibodies produced improvements in all clinically relevant measures and were considered to be equally effective. Furthermore, these improvements occurred after six months of therapy and were similar to the results obtained after 12 months.
Citation
Kayser MZ et al. Real-World Multicenter Experience with Mepolizumab and Benralizumab in the Treatment of Uncontrolled Severe Eosinophilic Asthma Over 12 Months. J Asthma Allergy 2021.
21st June 2021
The use of monoclonal antibodies have been shown to neutralise the COVID-19 virus in cell cultures and produce a significant reduction of viral load in vivo. A combination of two mAbs, casirivimab and imdevimab, which bind to separate parts of the COVID-19 receptor binding domain of the spike protein, are able to block entry of the virus into host cells. Since the mAbs bind to separate sites, there is reduced risk of antiviral failure. The use of this mAbs cocktail, known as REGEN-COV, has been shown to reduce the risk of hospitalisation or death among infected patients, in an outpatient setting. In addition, the manufacturer, Regeneron, has provided initial data which suggests that in hospitalised, seronegative patients, REGEN-COV reduced daily viral load. Whilst encouraging, no anti-viral therapy has been shown to reduce death among those hospitalised with COVID-19. This led the RECOVERY team to undertake a randomised trial of REGEN-COV among seropositive patients, hospitalised with COVID-19. For the trial, eligible patients (those with either clinically suspected or confirmed COVID-19 infection), were randomised in a 1:1:1 ratio to either standard care or standard care plus REGEN-COV or standard care and convalescent plasma. Among patients assigned to REGEN-COV, treatment consisted of a single dose (4g of both mAbs) in 250 ml saline infused over 60 minutes. The primary outcome of the trial was 28-day all-cause mortality and secondary outcomes included the time to discharge from hospital and in those not receiving mechanical ventilation at randomisation, a composite outcome of mechanical ventilation or death.
Findings
A total of 4839 patients were randomised to REGEN-COV and 4946 to usual care. The mean age of study participants was 61.9 years (37% female) and 78% of white ethnicity. However, the complete cohort included 3153 (32%) participants who were seronegative and 5272 (54%) who were seropositive. Among those allocated to REGEN-COV, 24% of seronegative patients and 30% of patients receiving usual care died within 28 days (rate ratio, RR = 0.80, 95% CI 0.70–0.91, p = 0.001).
In an analysis all the whole cohort (irrespective or serological status), 20% of those allocated to REGEN-COV and 21% of those given standard care died within 28 days (RR = 0.94, 95% CL 0.86 – 1.03, p = 0.17). Furthermore, among seronegative patients, progression to mechanical ventilation or death was lower among those receiving REGEN-COV compared to standard care (28% vs 32%).
In discussing their results, the authors noted that there was only an apparent benefit in seronegative patients receiving REGEN-COV. The suggested that the use of REGEN-COV should be restricted to seronegative patients.
Citation
RECOVERY Collaborative Group. Casirivimab and imdevimab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. MedRxiv 2021
IMPORTANT LINKS
MORE FROM COGORA
© Cogora 2025
Cogora Limited. 1 Giltspur Street, London EC1A 9DD
Registered in the United Kingdom. Reg. No. 2147432