






Systemic lupus erythematosus (SLE) is a multisystemic autoimmune disease, the spectrum of which covers a wide array of clinical and laboratory manifestations. The disease has considerable clinical and immunological heterogeneity; no two patients with SLE are exactly alike. The aetiology of SLE is thought to be multifactorial, with multiple genetic, epigenetic, hormonal, and immunopathological pathways being involved. SLE is characterised by the production of autoantibodies which leads to immune complex deposition, inflammation, and eventually, permanent organ damage. The course of SLE is largely unpredictable and characterised by periods of remission and disease exacerbation that could lead to progressive organ damage and dysfunction. It most commonly presents in women with a peak incidence between the ages of 15 and 40.1 However, SLE can affect all age groups, from infants to geriatric patients. Compared with the age- and sex-matched general population, SLE is associated with at least a five-fold increase in mortality.2
Accurate clinical assessment of the disease is desirable because SLE has a complex phenotype, a cumulative damage and a variable disease course with new organ system involvement even many years after diagnosis.
For all these reasons, the availability of measures for diagnosing, monitoring disease activity, and assessing tissue damage are all important and necessary in SLE management. Assessment of SLE3 can be divided into several components:
- diagnosis;
- monitoring;
- comorbidities;
- family planning.
Diagnosis
All patients suspected of having SLE should be referred to a rheumatologist or to a SLE specialist to confirm diagnosis and be involved in ongoing care. When considering a patient with a possible diagnosis of SLE, a detailed clinical history and examination is required in order to identify relevant clinical features. An early diagnosis of SLE could be challenging for many reasons: the absence of pathognomonic findings, the heterogeneity at onset and the time required for its full development. This peculiar disease pattern could explain the long delay reported between the onset of the first symptoms and the final diagnosis. In the last decades, however, the delay between clinical onset and diagnosis has been reduced, from more than 2 years in patients diagnosed during the eighties to nine months in those diagnosed in the last years.4 The Systemic Lupus International Collaborating Clinics (SLICC) set of classification criteria (Table 1)5 and the new American College of Rheumatology and European League Against Rheumatism (ACR/EULAR ) criteria (Table 2)6 may be helpful also considering the diagnosis; however, they do not cover all the possible clinical manifestations of SLE.
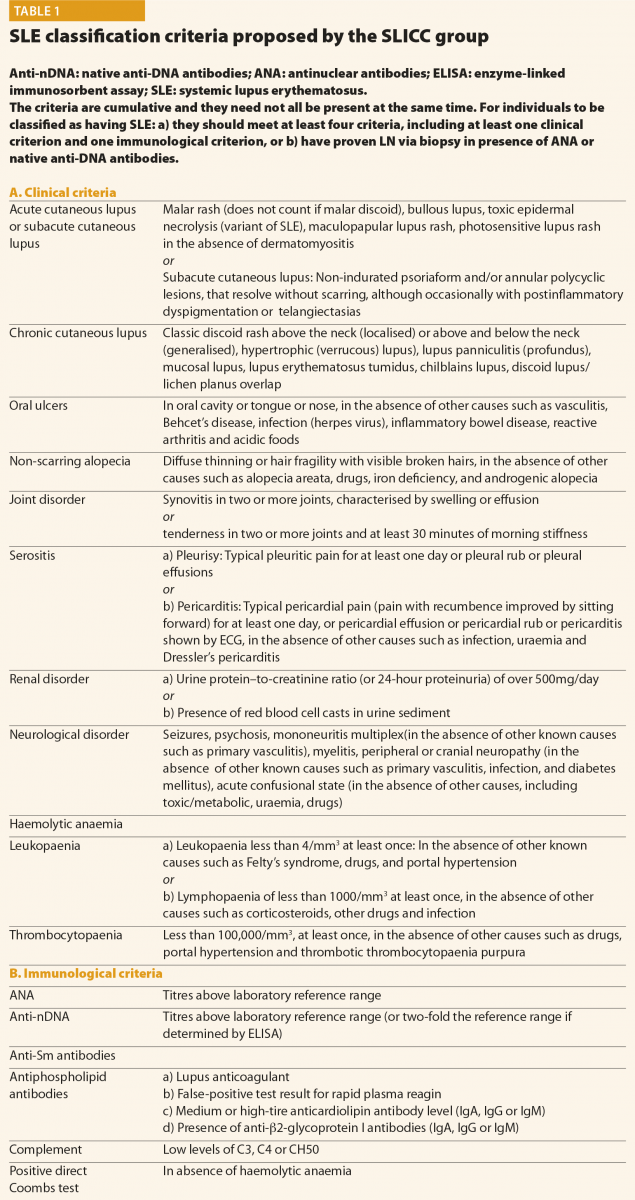
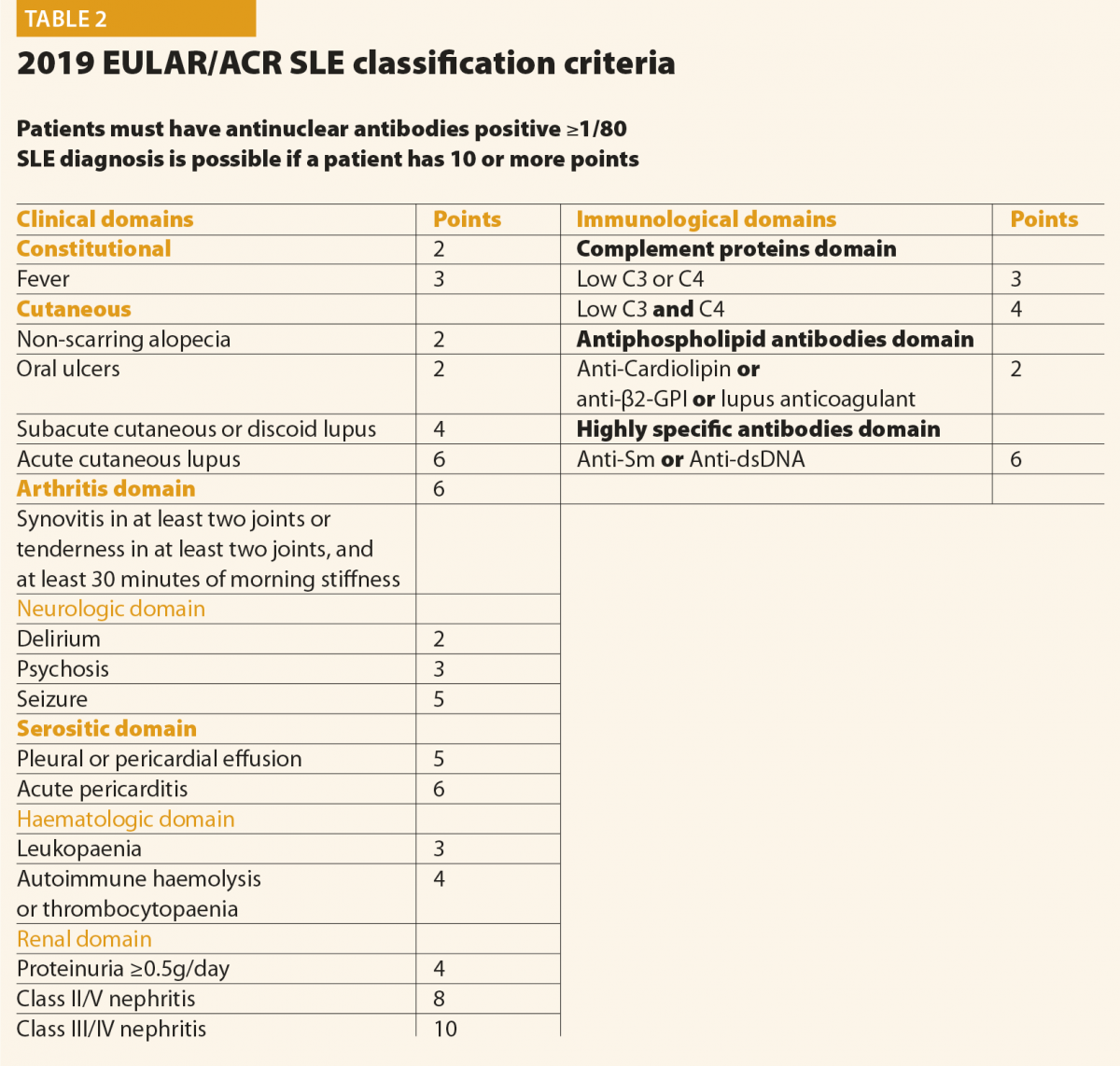
In conclusion, final diagnosis of SLE is a combination of clinical features and the presence of at least one relevant immunological abnormality and it still requires the meticulous clinical judgment of qualified physicians.
Screening questions should be employed to detect possible SLE manifestations in all systems of the body, particularly manifestations included in the classification criteria and others that are common in lupus patients, for example, fatigue, photosensitivity, skin lesions, arthralgias, alopecia and Raynaud’s phenomenon. Constitutional symptoms such as fatigue, fever, unintentional weight loss and lymphoadenopathy are common presenting symptoms of SLE. These symptoms, however, are not specific and other causes should be excluded, such as infection, malignancy, endocrinopathy, other connective tissue diseases, depression and fibromyalgia. In addition, environmental triggers such as exposure to ultraviolet radiation, infection, or the use of certain medications, should be identified, if possible. At the moment of the diagnosis, a complete clinical and immunological evaluation should be performed, as reported in Table 3. Possible kidney or neurological involvement should always be ruled out, because involvement of these organs is considered as a severe SLE manifestation.
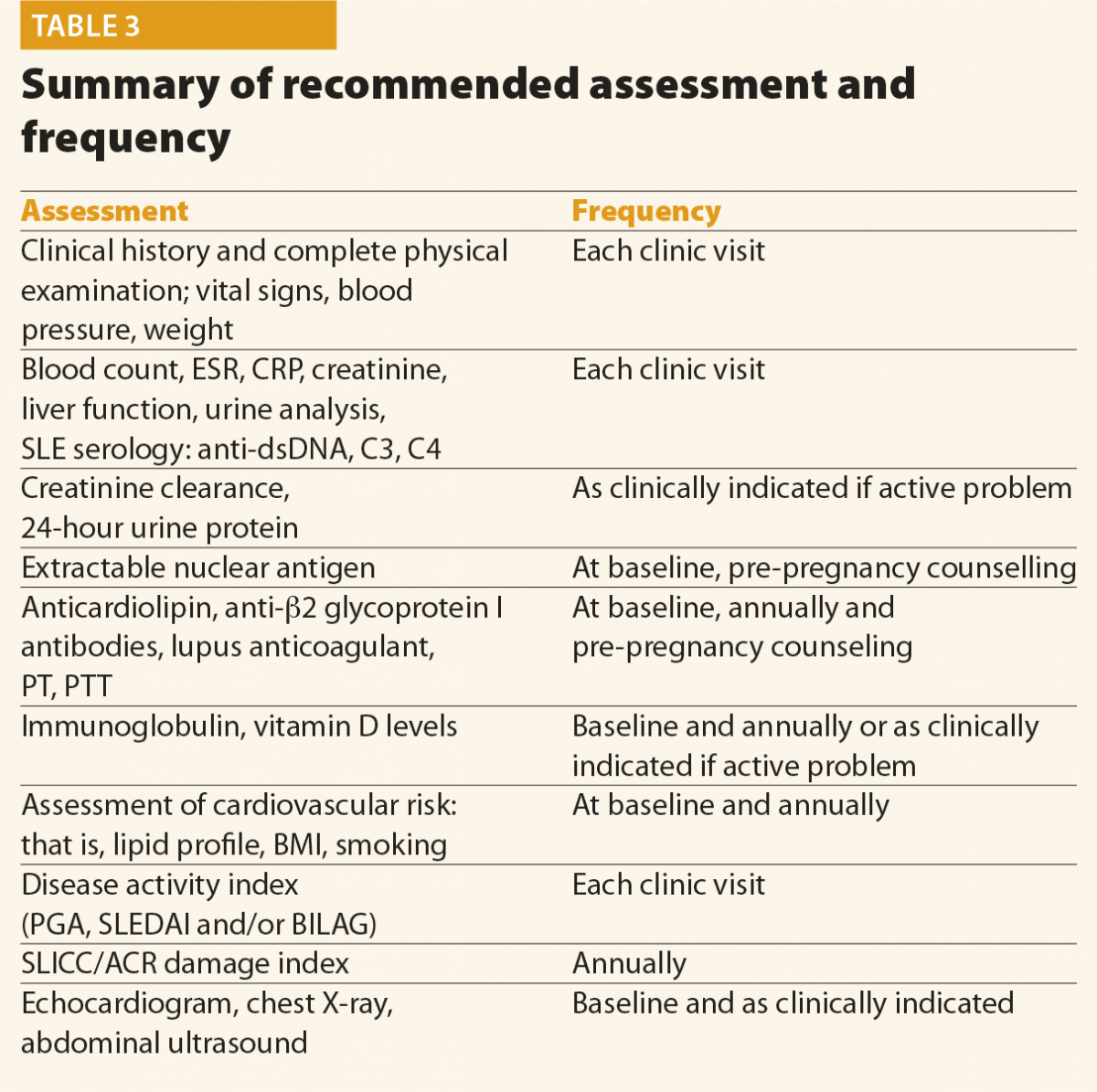
Monitoring
No data are available in the literature to suggest an optimal frequency of clinical and laboratory assessment in patients with SLE. Table 3 summarises the assessment and monitoring of patients with SLE.3
The frequency of the follow-up visits should be based on the activity and severity of the disease, its complications and evolution. In patients with inactive disease, without organ damage and comorbidities, the EULAR recommends that clinical and laboratory examinations should be carried out every 6–12 months.7 Patients with active or more severe disease, with complications related to the treatment, as well as when immunosuppressant treatment is introduced or reduced, will need more frequent control.
How to monitor a SLE patient
SLE is a very heterogeneous disease and its activity fluctuates over time. This variability means that patients with SLE require standardised and objective monitoring of the disease, with validated instruments to determine the degree of activity.
The use of an activity index is also desirable in routine clinical practice as a way of guiding therapeutic decisions as objectively as possible. Moreover, clinicians have to be able to distinguish disease activity from chronic damage, infection and other comorbid disease, including drug side effects.
Many indices have been proposed and all of these have been proven to be valid to measure the activity of SLE and they are also able to predict damage and mortality.8 In addition to the Physicians’ Global Assessment (an estimate of activity rated on a 0 to 3 visual analogue scale), the most common measures used include the SLE Disease Activity Index (SLEDAI), the British Isles Lupus Assessment Group (BILAG), the Systemic Lupus Activity Measure (SLAM), the Lupus Activity Index (LAI), and the European Consensus Lupus Activity Measurement (ECLAM).
The SLEDAI index and its latest version SLEDAI-2K9 is one of the most commonly used global disease activity measure in clinical practice and clinical trials (Table 4). The score has demonstrated validity, reliability, and sensitivity to change in several observational studies. The practical applicability of SLEDAI-2K in clinical settings, its ease of administration (less than 10 minutes to completed), and its simplicity in scoring are fundamental properties. The SLEDAI-2K includes the evaluation of 24 weighted objective variables (16 clinical and 8 laboratory; Table 4). A manifestation is recorded if it is present over the past 10 days. The sum of the items, identified in a single patient, corresponds to the disease activity. Concerning the laboratory items, the SLEDAI includes the determination of complement levels and of anti-dsDNA antibodies. The main limitation is that the SLEDAI-2K is not able to capture improvements or worsenings within an organ system, and it does not include severity. By contrast, the BILAG score10 (Figure 1) refers to disease activity in nine organ systems
in the previous 30 days. For each item included, a score is assigned according to the degree of disease activity. The BILAG is the most comprehensive activity index, and it has the power to detect changes in the activity of the disease, therefore it is frequently used in randomised clinical trials. However, it is longer than SLEDAI to complete (almost one hour), it requires several laboratory parameters and
a dedicated software. In conclusion, it is not feasible in everyday practice.
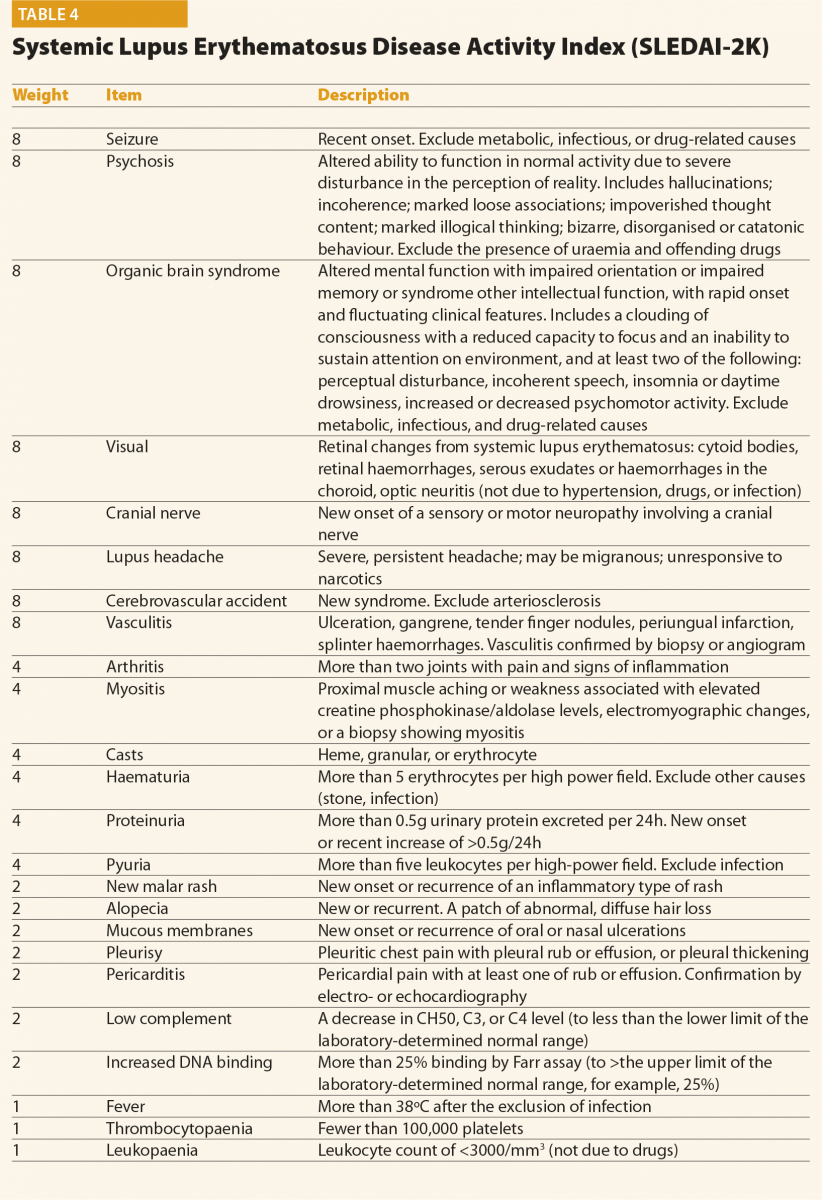

Other than disease activity, damage caused by the disease also has to be assessed. Since 1996, a damage index proposed by Systemic Lupus International Collaborating Clinics has been used – the SLICC/ACR Damage Index (SLICC/ACR DI).11 This index includes persistent damage related to SLE per se (present for more than six months), specific comorbidities associated to the autoimmune disease, and features that are often due to toxicity related to treatments. Higher damage index scores early in disease have been associated with a poor prognosis and with increased mortality.12 Thus, the SLICC/ACR DI complements other measures of disease activity described above, and it is an important outcome measure. It is usually completed (or updated) yearly.
Lifestyle guidance
Finally, lifestyle modifications should be discussed. All SLE patients should stop smoking, not just because of the effect that smoking has
on the activity of the disease and quality of life,13 but also because of its causal association with the increase in risk of cardiovascular disease, infection and cancer. Regular physical exercise in people with stable SLE with low to moderate disease activity should be recommended in order to avoid weight gain expecially in those patients that are taking corticosteroids.
Adequate photoprotection should be advised in all SLE patients, irrespective of the presence of skin manifestations.14 Broad spectrum photoprotectors with high solar photoprotection index should be applied in adequate quantity before exposure and frequently reapplied and
the use of hat and long-sleeved clothing should be advised.
Comorbidities
Patients with SLE are at increased risk of comorbidities such as infection, thrombotic events, premature cardiovascular and peripheral vascular disease and osteoporosis. These aspects should be assessed at diagnosis and regularly during follow-up. The frequency of the proposed assessment is reported in Table 3.
Antiphospholipid antibodies, thrombosis and cardiovascular risk
Antiphospholipid antibodies (aPL) are considered a risk factor of thrombosis, both in the presence and in the absence of a concomitant autoimmune disease such as SLE.15
It is very important to remember that all the three test, namely lupus anticoagulant (LA), anticardiolipin (aCL) and anti-β2glycoprotein
I antibodies (anti-β2-GPI) should be determined at baseline and regularly during the follow-up because only their complete evaluation allow to assess the risk profile. It has been demonstrated that the combinations of aPL generally increase the risk of thrombosis, and the triple positivity is associated with the greatest likelihood of thromboembolic events.16
The decision for the use of primary prophylaxis in aPL carriers remains challenging: patients at ‘high risk’ (that is, triple positivity, high titres of autoantibodies) should be treated with low-dose aspirin, whereas in patients considered at ‘low risk’, the decision should be made taking into consideration also the classical cardiovascular risk factors, age and the risk of bleeding.
Concerning the secondary prevention, for both unprovoked venous thromboses and arterial thrombosis, indefinite anticoagulation is recommended.17
The use of novel oral anticoagulants for secondary prevention should be avoided, especially in patients with triple aPL positivity.18
It is well known that SLE patients present an increase in risk of cardiac events19 and coronary disease compared with the rest of the population. The risk that a young female SLE patient develops cardiovascular events was reported 30–50-times higher than that of the same age healthy control. This excess of risk is not entirely explained by classical risk factors, but rather, other factors related to the actual disease may also be involved, such as chronic systemic inflammation or treatment with glucocorticoids.19,20
Traditional risk algorithms (for example, Framingham) show only marginal differences between SLE and controls, and thus underestimate cardiovascular disease risk of SLE patients. A new risk score to predict the development of cardiovascular disease has been reported (QRISK3)21 where SLE is considered as a risk factor. The use of QRISK3 was able to capture significantly more patients with SLE
with an elevated 10-year risk of developing cardiovascular disease compared with Framingham.22 The European Soiety for Cardiology includes SLE in the population group with increased risk of suffering cardiovascular disease,23 for whom it recommends a target LDL level lower than 2.5mmol/l (96mg/dl).
Infection
Patients with SLE are at high risk for infections because of the underlying immune aberrations and the prolonged use of immunosuppressive treatments.24,25 Protection against infections should focus both on primary prevention, as well as promp recognition and treatment.
Patients with SLE should receive vaccinations as proposed by EULAR in the recommendations for vaccination of patients with autoimmune rheumatic diseases.24 Seasonal influenza and pneumococcal vaccination are strongly recommended for patients with SLE, preferably during stable disease.24,26 Live attenuated vaccines are not recommended in patients chronically treated with immunosuppressant therapies because of the risk of disseminated infections.
Osteoporosis
Osteopaenia and osteoporosis are frequent comorbidities in SLE patients and the disease itself represents an independent risk factor for low bone mineral density (BMD), but there are additional risk factors that may concur, such as therapy with glucocorticoids and the high prevalence of vitamin D insufficiency or deficiency,27 favoured also by the doctor’s prescribed lifestyle. BMD score should be evaluated at disease onset, and the ongoing EULAR recommendations suggest the supplementation of elemental calcium and cholecalciferol in all patients chronically treated with low-medium dosage of corticosteroids.28
In case of vitamin D deficiency, higher dosage should be initially prescribed and then lowered to a standard dosage of cholecalciferol (600–800IU/day). In patients with SLE aged >40 years, who have a moderate to high risk of a major osteoporotic (>10%) or hip fracture (>1%) within 10 years (as assessed by the fracture risk assessment tool), antiresorptive therapy is recommended if there are no contraindications.
Family planning
Clinicians involved in the care of SLE patients should frequently discuss the topic of family planning with all patients of childbearing age. Family planning and contraception should be a topic of discussion from the early phases of the disease.
In the past, SLE was considered to be an absolute contraindication to pregnancy, but maternal and foetal outcomes in these women have greatly improved thanks to a correct timing of pregnancy, close monitoring, multidisciplinary management and an increased knowledge about the medications that can be used during pregnancy and breastfeeding.29,30
Ideally, all patients with SLE who wish to conceive should have preconception counselling; in fact, planned pregnancies have demonstrated better maternal and foetal outcomes. Systemic activity of SLE must be assessed, with careful attention to renal involvement. A recent flare is a risk factor for recurrence during pregnancy; therefore a pregnancy should be planned after
at least six consecutive months of stable disease. During the preconceptional visit, the SLE specialist should:
- perform a risk assessment: other than the common epidemiological risk factors that are relevant to any pregnancy, the physician should revise previous obstetric history, assess activity (SLEDAI-2K and/or BILAG) and irreversible damage (SLICC-SDI DI);
- complete laboratory testing, with particular attention to aPL status, presence of anti-Ro/SSA or LA/SSB antibodies
- review therapies, as immunosuppression, antihypertensives and anticoagulation.
At the same time, patients need to be evaluated by an experienced obstetrician who will perform the investigation required for all the women who wish to get pregnant.
Once pregnancy is confirmed, monthly visits are usually indicated (or even more frequently)
if the disease is not controlled.
Special monitoring is dedicated to women with Ro/SSA and/or La/SSB antibody positivity; these patients should be informed about the risk of neonatal lupus and congenital heart block, a severe complication that occurs in 0.7–2%31,32 of women with these autoantibodies.
Conclusions
Physicians involved in the care of patients affected by SLE need easy to use, replicable and practical evaluation tools. These can help in all disease phases, from diagnosis through to the occurrence of flares, comorbidities, or even pregnancy. SLE shows a variable and complex phenotype; therefore a precise assessment is needed to administer the treatment able to target the index manifestation. Only by following this strategy will we be able to control the disease, reaching remission or low disease activity,26 which are essential in improving
the morbidity, mortality and quality of life in these patients.
References
1 Wahren-Herlenius M et al. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet 2013;382:819–31.
2 Mok CC et al. Life expectancy, standardized mortality ratios, and causes of death in six rheumatic diseases in Hong Kong, China. Arthritis Rheum 2011;63:1182–9.
3 Mosca M et al. European League Against Rheumatism recommendations for monitoring patients with systemic lupus erythematosus in clinical practice and in observational studies. Ann Rheum Dis 2010;69:1269–74.
4 Doria A et al. SLE diagnosis and treatment: when early is early. Autoimmun Rev 2010;10:55–60.
5 Petri M, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012;64:2677–86.
6 Aringer M et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus.Ann Rheum Dis 2019;78(9):1151–9.
7 Mosca M et al. Development of quality indicators to evaluate the monitoring of SLE patients in routine clinical practice. Autoimmun Rev 2011;10:383–8.
8 Griffiths B et al. Assessment of patients with systemic lupus erythematosus and the use of lupus disease activity indices. Best Pract Res Clin Rheumatol 2005; 19:685–708.
9 Glad-man DD et al. Systemic Lupus Erythematosus Disease Activity Index 2000. J Rheumatol 2002;29:288–91.
10 Hay EM et al. The BILAG index: a reliable and valid instrument for measuring clinical disease activity in SLE. Q J Med 1993;86:447–58.
11 Gladmand D et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum 1996;39:363–9.
12 Rahman P et al. Early damage as measured by the SLICC/ACR damage index is a predictor of mortality in systemic lupus erythematosus. Lupus 2001;10(2):93–6.
13 Ghaussy NO et al. Cigarette smoking and disease activity in systemic lupus erythematosus. J Rheumatol 2003;30:1215–21.
14 Vilá L et al. Association of sunlight exposure and photoprotection measures with clinical outcome in systemic lupus erythematosus. P R Health Sci J 1999;18:89–94.
15 Miyakis S, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4: 295–306.
16 Chighizola CB et al. The treatment of anti-phospholipid syndrome: a comprehensive clinical approach. J Autoimmun 2018;90:1–27.
17 Tektonidou MG et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann Rheum Dis 2019; Epub ahead of print doi:10.1136/annrheumdis-2019-215213.
18 Pengo V et al. Rivaroxaban vs warfarin in high-risk patients with antiphospholipid syndrome. Blood 2018;132:1365–71.
19 Magder LS, Petri M. Incidence of and risk factors for adverse cardiovascular events among patients with systemic lupus erythematosus. Am J Epidemiol 2012; 176: 708-19.
20 Esdaile JM et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum 2001;44:2331–7.
21 Hippisley-Cox J et al. Development and validation of QRISK3 risk prediction algorithms to estimate future risk of cardiovascular disease: prospective cohort study. BMJ 2017;357:j2099
22 Edwards N et al. QRISK3 improves detection of cardiovascular disease risk in patients with systemic lupus erythematosus. Lupus Sci Med 2018;5:e000272.
23 Perk J et al. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012): The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice. Atherosclerosis 2012;223:1–68.
24 Elkayam O et al. Update of EULAR recommendations for vaccination of patients with autoimmune inflammatory rheumatic diseases. Ann Rheum Dis 2018;77.
25 Rúa-Figueroa I et al. Incidence, associated factors and clinical impact of severe infections in a large, multicentric cohort of patients with systemic lupus erythematosus. Semin Arthritis Rheum 2017;47:38–45.
26 Fanouriakis A et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis 2019;78(6):736–45.
27 Almehed K et al. Prevalence and risk factors of osteoporosis in female SLE patients-extended report. Rheumatology (Oxford) 2007;46:1185–1190.
28 Grossman JM et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2010;62:1515–26.
29 Andreoli L et al. EULAR recommendations for women’s health and the management of family planning, assisted reproduction, pregnancy and menopause in patients with systemic lupus erythematosus and/or antiphospholipid syndrome. Ann Rheum Dis 2017;76(3):476–85.
30 Götestam Skorpen C et al. The EULAR points to consider for use of antirheumatic drugs before pregnancy, and during pregnancy and lactation. Ann Rheum Dis 2016;75(5):
795–810.
31 Brucato A et al. Risk of congenital complete heart block in newborns of mothers with anti-Ro/SSA antibodies detected by counterimmunoelectrophoresis: a prospective study of 100 women. Arthritis Rheum 2001;44:1832–5.
32 Fredi M et al. First report of the Italian Registry on immune-mediated congenital heart block (Lu.Ne registry). Frontiers Immunology 2019;6:11.
